Rev’s Transcript Library
Explore our extensive collection of free transcripts from political figures and public events. Journalists, students, researchers, and the general public can explore transcripts of speeches, debates, congressional hearings, press conferences, interviews, podcasts, and more.

Blanche Confirmation Day Two
Todd Blanche confirmation hearing for Attorney General, day two. Read the transcript here.

Blanche Confirmation Day One
Todd Blanche testifies at confirmation hearing to be Attorney General, day one. Read the transcript here.

Trump Address to the Nation 7/16/26
Donald Trump speaks to the nation on election integrity claims. Read the transcript here.

UT v. Tyler Robinson Preliminary Hearing Day 5
Day 5 of the UT v. Tyler Robinson Preliminary Hearing. Read the transcript here.
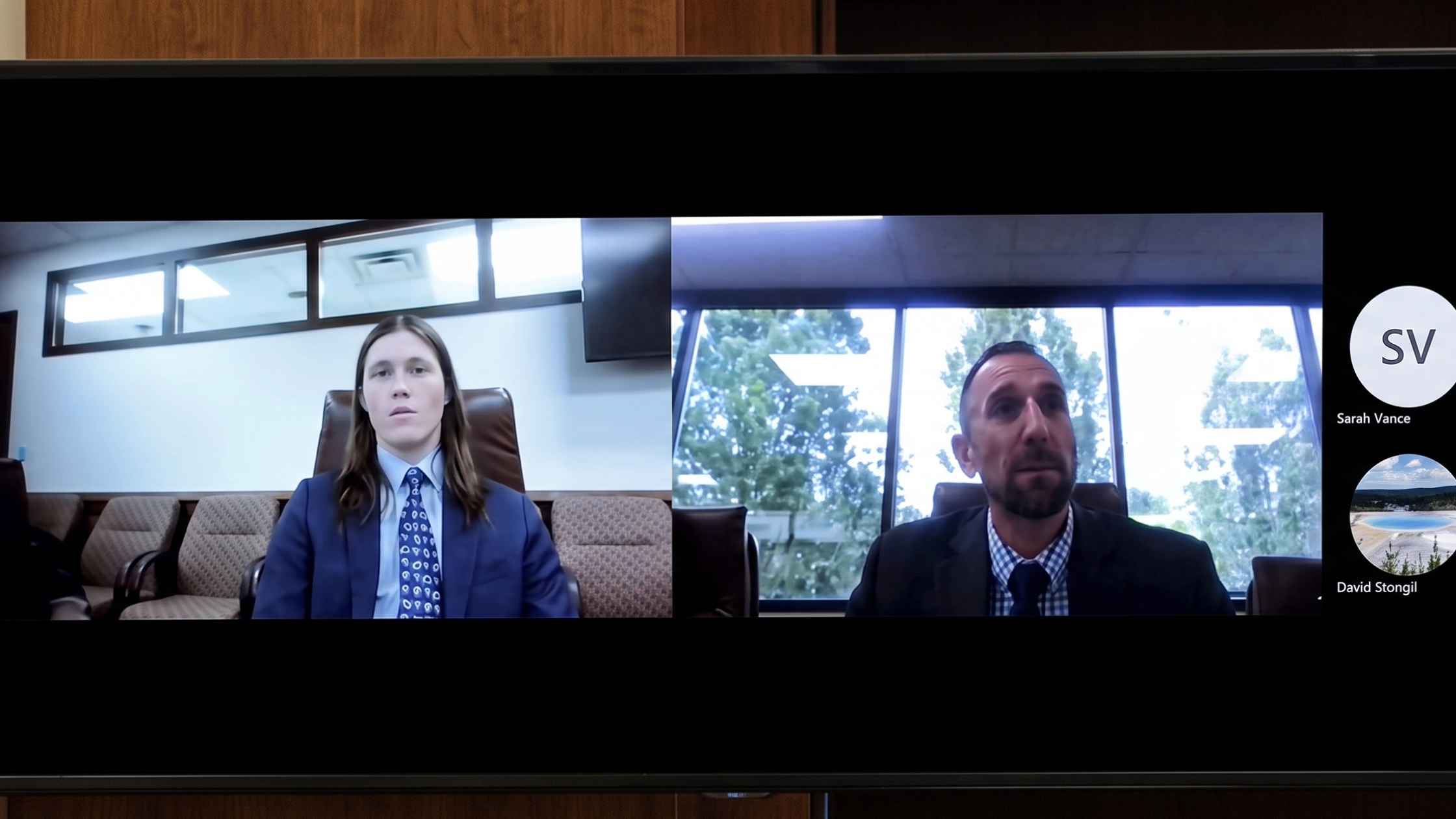
UT v. Tyler Robinson Preliminary Hearing Day 4
Day 4 of the UT v. Tyler Robinson Preliminary Hearing. Read the transcript here.

UT v. Tyler Robinson Preliminary Hearing Day 3
Day 3 of the UT v. Tyler Robinson Preliminary Hearing. Read the transcript here.

Update on High-Rise Collapse Risk
Zohran Mamdani gives an update as Manhattan buildings are evacuated over high-rise collapse risk. Read the transcript here.

78th Emmy Awards Nominations
Emmy winners Liza Colón-Zayas and Jeff Hiller announce the nominees for the 78th Emmy Awards. Read the transcript here.

UT v. Tyler Robinson Preliminary Hearing Day 2
Day 2 of the UT v. Tyler Robinson Preliminary Hearing. Read the transcript here.

Trump Meets Zelenskiy on Sidelines of NATO Summit
Donald Trump meets Ukrainian President Volodymyr Zelenskyy on the sidelines of a NATO summit in Ankara, Turkey. Read the transcript here.

UT v. Tyler Robinson Preliminary Hearing Day 1
Charlie Kirk's accused killer, Tyler Robinson, is in court as prosecutors begin a preliminary hearing. Read the transcript here.

America 250 Keynote Address
Donald Trump delivers the keynote address at America 250. Read the transcript here.
Subscribe to The Rev Blog
Sign up to get Rev content delivered straight to your inbox.